Elfride and several lab members went to Salt Lake City in Utah for the annual ARVO meeting. Elfride chaired a session on inherited eye disease and our lab had several oral presentations and posters to present.
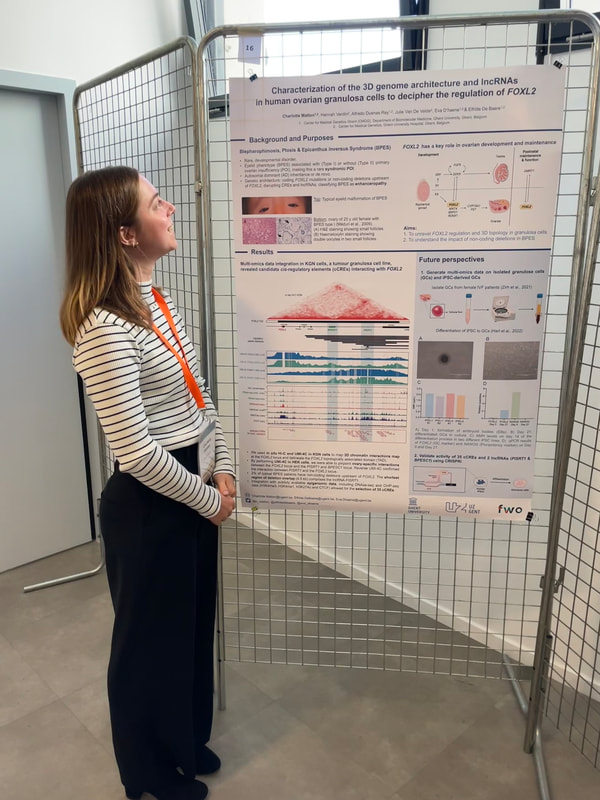
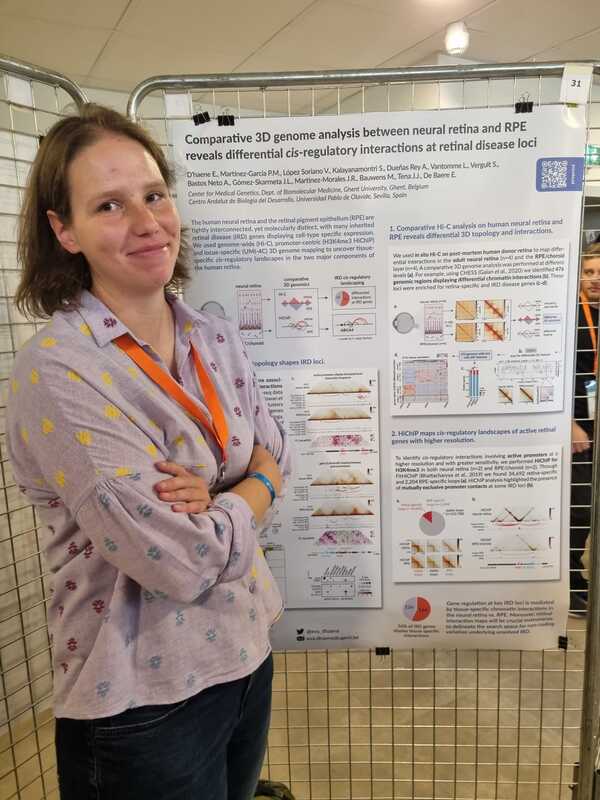
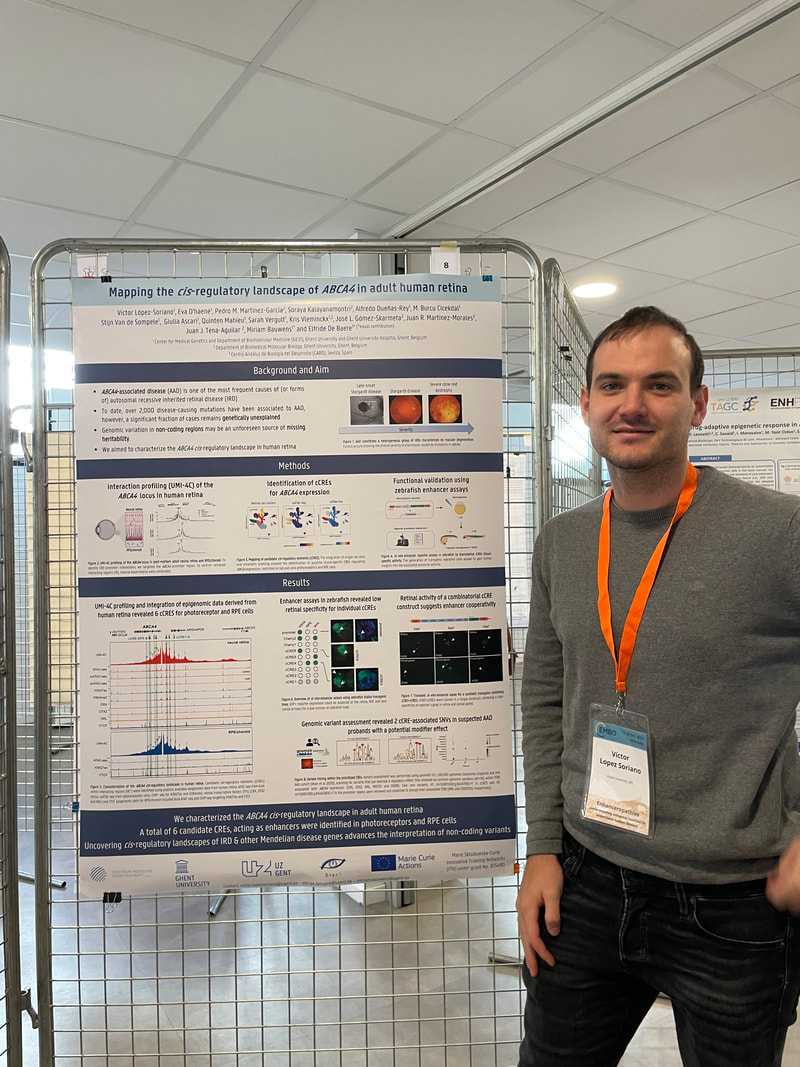
From 17-20th of October, 4 members of the De Baere Lab team attended the EMBO workshop on enhanceropathies: understanding enhancer function to understand human diseases, in Marseille (France). Lieselot presented a poster on one of the causes of North Carolina Macular Dystrophy (NCMD), namely non-coding duplications near IRX1. Chromatin interaction profiling of this region uncovered 10 candidate cis-regulatory elements (cCREs). Her poster details how she will investigate the underlying disease mechanisms of NCMD, a retinal enhanceropathy. Charlotte presented a poster on her research deciphering the cis-regulatory region of FOXL2, a key factor in ovarian development. By combining her self-generated Hi-C and UMI-4C data in granulosa cells with publicly available OMICS data, she was able to identify 35 cCREs and 2 lncRNAs that will be validated using CRISPRi. Eva presented a poster on a comparative 3D genome analysis between neural retina and RPE and the implications for the regulation of retinal disease genes. Lastly, also Victor presented a poster where he unravels the cis-regulatory landscape of ABCA4, in adult human retina.
NEW PAPER ALERT!
By Alfredo Duenas Rey & Victor Lopez Soriano Cross-species genome comparisons have revealed a substantial number of ultraconserved non-coding elements (UCNEs). Several of these elements have proved to be essential tissue- and cell type-specific cis-regulators of developmental gene expression. Here, we characterized a set of UCNEs as candidate CREs (cCREs) during retinal development and evaluated the contribution of their genomic variation to rare eye diseases, for which pathogenic non-coding variants are emerging. Integration of bulk and single-cell retinal multi-omics data revealed 594 genes under potential cis-regulatory control of UCNEs, of which 45 are implicated in rare eye disease. Mining of candidate cis-regulatory UCNEs in WGS data derived from the rare eye disease cohort of Genomics England revealed 178 ultrarare variants within 84 UCNEs associated with 29 disease genes. Overall, we provide a comprehensive annotation of ultraconserved non-coding regions acting as cCREs during retinal development which can be targets of non-coding variation underlying rare eye diseases.
Prof. Elfride De Baere, received a generous fund granted by the King Baudouin Foundation last week. This grant will be used for further research studies in our research group on functional genomics in the retina.  Dr. Anouk Georges (MD, PhD) joined us for a very interesting seminar about comparing the transcriptome of developing native and iPS-derived mouse retinae by scRNA-seq & ATAC-seq.  NEW PAPER ALERT!
By Alexandre Segers RNA-sequencing (RNA-seq) is increasingly used to diagnose patients with rare diseases by prioritising genes with aberrant expression and/or splicing. State-of-the-art methods for detecting aberrant expression and splicing, however, are extremely slow. The latter, also discard much information because they only use junction reads to infer aberrant splicing. In this contribution, we show that replacing the offset for library size unlocks conventional bulk RNA-seq workflows for fast and scalable differential usage, aberrant splicing and expression analyses. Our method, saseR, is several orders of magnitude faster than the state-of-the-art methods and dramatically outperforms these in terms of sensitivity and specificity for aberrant splicing, while being on par with these inferring differential usage and aberrant expression. Finally, our framework is also very flexible and can be used for all applications that involve the analysis of proportions of short- or long RNA-seq read counts.
|