|
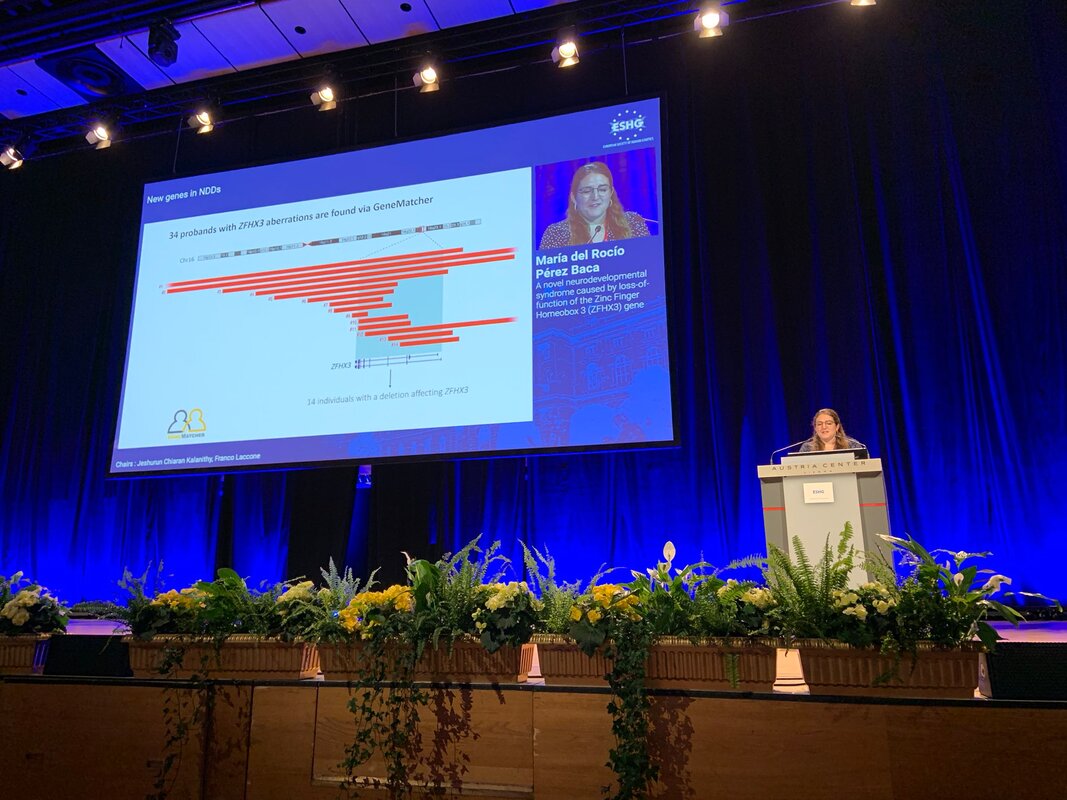
Three fabulous keynote speakers were invited to the RARE-MED symposium covering non-coding variantion, Nicky Whiffin (Oxford Universoty), functional genomics, Musa Mhlanga (Radboud University, Niijmegen, The Netherlands) and RNA therapy of Neurological Diseases, Willeke Van Roon-Mom (leiden University Medical Center & Dutch center for RNA therapeutics, Leiden, The Netherlands). Apart from this, several members of our team presented their research (posters and oral presentations). Special acknowledgement to our team member Eline Van Vooren who won the price for Best Poster with her research on RPE65 VUS assessment. We are looking forward to next year's edition! No less than six lab members had an oral talk at the 2022 ESHG meeting. Sarah Vergult and Eva D'Haene presented on Structural variants disrupt a critical regulatory region downstream of FOXG1 and Mapping the 3D genome of the human retina and its role in retinal disease, respectively. Stijn Vandesompele's talk was titled Dual molecular effects of constitutional SF3B2 variants cause a novel dominant spliceosomopathy displaying retinitis pigmentosa or developmental skeletal anomalies and Rocio discussed A novel neurodevelopmental syndrome caused by loss-of-function of the Zinc Finger Homeobox 3 (ZFHX3) gene. Victor López Soriano had a talk on An integrated approach using multi-omics data to dissect cis-regulatory role of ultraconserved non-coding elements (UCNRE) in human retina in the same session as Alfredo, who spoke Identification and charaterization of a novel retina-specific lncRNA upstream ABCA4 with a potential role in ABCA4-associated inherited retinal disease. Congrats to all on the very nice presentations! Miriam Bauwens had a poster presentation.
Three Lab De Baere members had an oral presentation and six had a poster presentation at the 22th Belgian Society for Human Genetics meeting in Bruges. Congrats to Eva, Burcu and Rocio for their oral presentations on Mapping the 3D genome of the human retina and its role in retinal disease, Single-cell transcriptional dynamics and in vivo enhancer assays provide insight into gene regulatory networks of PRDM13 and IRX1 implicated in North Carolina macular dystrophy and A novel neurodevelopmental syndrome caused by loss-of-function of the Zinc Finger Homeobox 3 (ZFHX3) gene, respectively. Posters by Miriam, Marta, Alfredo, Charlotte, Marjolein, Victor were visable during the poster sessions. We also had fun during the conference dinner, party and especially during the suprise act by science comedian Lieven Scheiren.
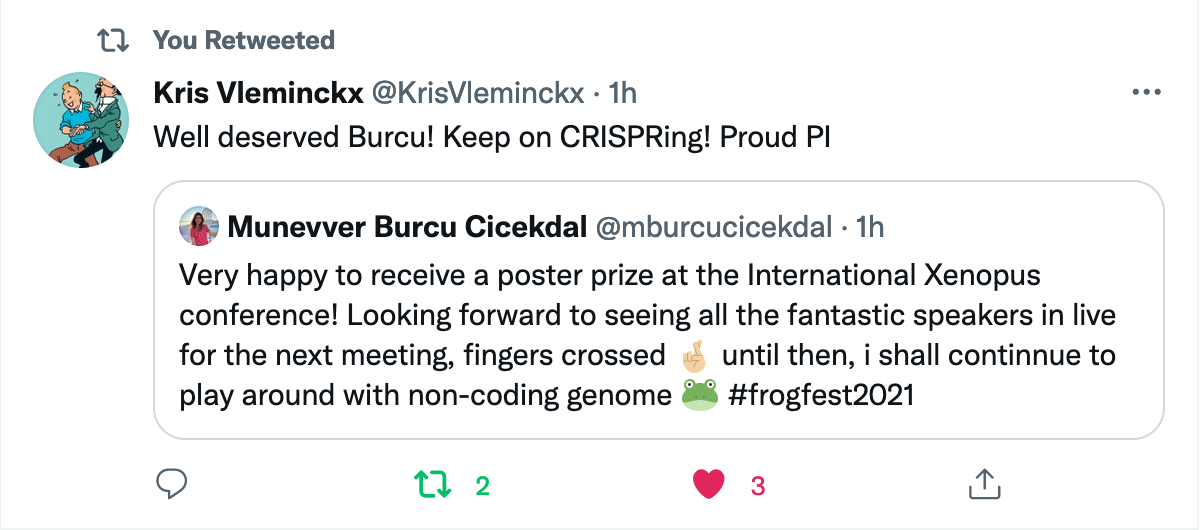
The European Vision Institute has announced its list of top women in European Vision Research and ophthalmology for 2021. Elfride De Baere was granted a place on the list. Congratulations Elfride  Münevver BurcU Cicekdal won the poster prize at the 18th international xenopus conference 202130/8/2021
Rare-med walks, runs, bikes to support rare diseases! Support our research on rare diseases!22/2/2021
We would like to invite you to the online kick-off symposium of the RARE-MED consortium on November 27, 2020, from 14-17.30 p.m. Registration is free of charge and can be done here. Please register if you wish to attend, as only registered people will receive a link for the symposium. The symposium program is displayed below. RARE-MED is a multidisciplinary UGent consortium for basic and translational research on precision medicine for rare diseases, to address missing heritability using systems genetics and functional genomics, to facilitate disease modelling using CRISPR/Cas9-mediated genome editing of aquatic model organisms (zebrafish, Xenopus) and of cellular systems, to introduce new gene therapies based on antisense oligonucleotide- or CRISPR/Cas9-based genome editing. In the context of this 21ZAP consortium, 3 research professors were appointed: Prof. dr. Sarah Vergult, prof. dr. Kris Vleminckx, and prof. dr. Frauke Coppieters. 
Giulia Ascari virtually defended her PhD thesis on June 18 2020. Congratulations to our doctor, whose favorite genes are RCBTB1 & CEP78. On the 27th of April 2020, Marlies Saelaert virtually defended her PhD thesis. This was an unforgettable experience. Congratulations! Our new team member, Karolien Aelbrecht, virtually defended successfully her PhD thesis on April 28 2020. Well done, congratulations Karolien! |